Viral cell-entry mechanisms
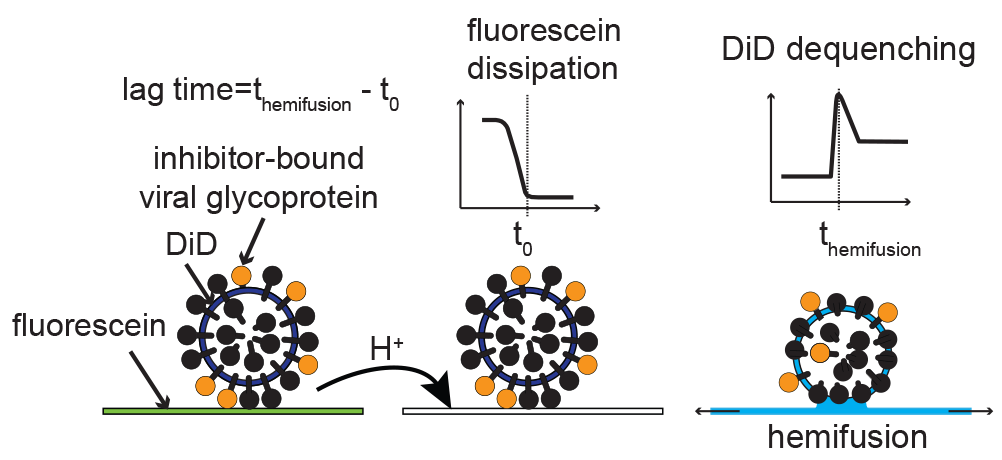
Viruses initiate infection by delivering the genomes from within the viral capsid or a membrane envelope to the target cell. Enveloped viruses accomplish this by fusing/merging their membrane with a cellular membrane. Membrane fusion is mediated by the viral surface glycoproteins which undergo a series of large-scale conformational changes that allow them to first insert in the target membrane then bring the viral and cellular membranes together. By measuring the fusion kinetics of individual virus particles and their mutants in the presence or absence of antibodies or inhibitors, we have uncovered the detailed mechanism of membrane fusion by influenza virus. Influenza cell-entry glycoprotein hemagglutinin (HA) is present at a high density on virus particles. Three to five HAs that insert next to each other cooperate to bring about membrane fusion. However, the steps leading to HA insertion are stochastic (noncooperative, random), so substoichiometic (nonsaturating) antibody or inhibitor amounts prevent fusion by the majority of particles.
A further consequence of the mechanism of membrane fusion we uncovered is that really large particles are virtually impossible to inhibit. Tens of micrometers-long viral filaments require greater than 90% of HAs to be inactivated for any reduction in the fusion yield. This insight led to a model where the pre-existing diversity in virus particle size in pleomorphic (mixed shape) viruses enables viral adaptation to cell-entry pressure in a way that does not require an initiating genetic change. We are asking: 1) How does virus particle shape influence viral adaptation to cell-entry pressure? 2) What is the mechanism of membrane fusion by other pleomorphic viruses (e.g. Ebola virus)? 3) What role do other viral structural features (e.g. glycoprotein density) play in the mechanism of membrane fusion? 4) What role do the interactions between viral glycoproteins and cellular receptors play in membrane fusion?
Viral assembly mechanisms
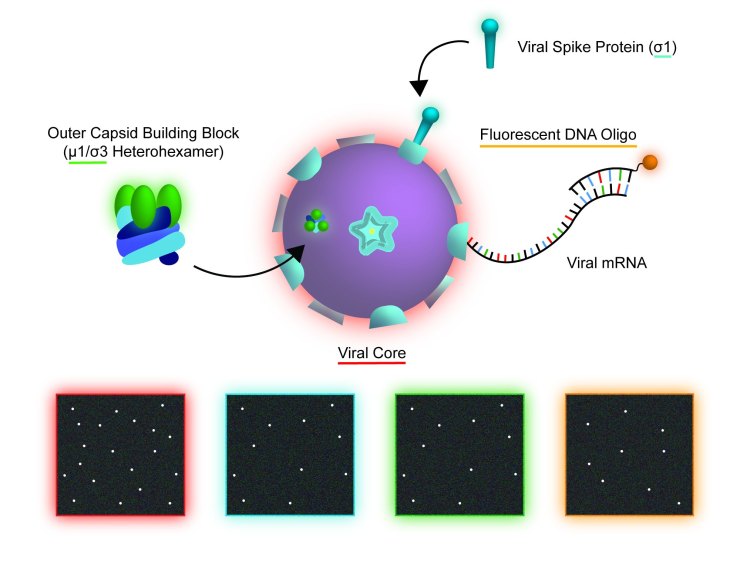
We have established a TIRF-based single-particle platform for quantitative real-time imaging of reovirus assembly, disassembly, and transcription. Reovirus virion consists of two concentric icosahedral protein shells. The inner, core particle encloses the genome and serves as the transcriptional unit in the infected cell cytoplasm. The outer capsid delivers the core across the cellular membrane to initiate infection. Disassembly of the outer capsid activates cores for transcription, and its assembly around cores represses transcription. In our experiment, fluorescently labeled cores are immobilized on microscope coverslips. Fluorescently tagged oligonucleotides detect nascent viral transcripts. Fluorescently tagged outer-capsid components assemble around immobilized cores and repress transcription. We are working toward a quantitative model of reovirus outer-capsid assembly and transcriptional regulation.
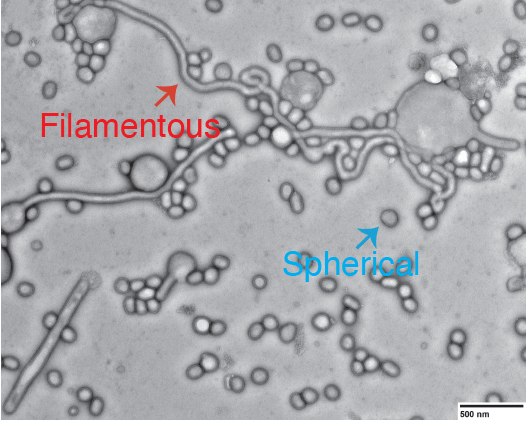
In addition to studying the assembly of regularly shaped, icosahedral virions, we are interested in mechanisms of assembly of irregularly shaped, pleomorphic viruses. For this, we have developed a novel, high-throughput assay enabling quantification of virus particle size distributions directly in the medium around infected cells. We are asking: 1) What is the mechanism of pleomorphic virion assembly? 2) How is the shape of pleomorphic virions regulated? 3) What are the viral and cellular determinants of viral assembly outcomes? 4) How do viruses with segmented genomes ensure efficient packaging of the full genome complement?
Viral adaptation – crosstalk between phenotypic and genetic diversity
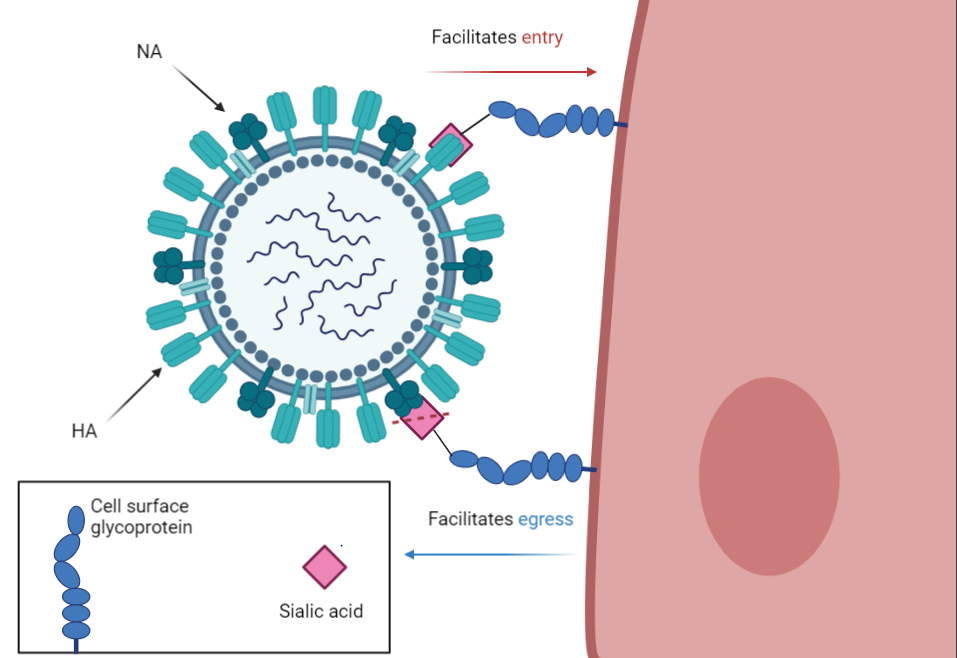
The main focus of the immune response against influenza virus are two glycoproteins on the virion surface, hemagglutinin (HA) and neuraminidase (NA). HA and NA have opposing functions: HA attaches virions to cells to initiate infection by binding to terminal sialic-acid residues on glycoproteins or glycolipids; NA frees newly assembled virions from the infected-cell surfaces by cleaving terminal sialic-acid residues from the cell and HA. Antibodies to HA and NA protect from influenza pathogenesis, but drive immune escape resulting from mutations in their epitopes. Notably, HA and NA functions co-evolve in ways that both constrain and enable adaptation. For example, stronger receptor binding by HA has to be accompanied by greater NA activity to retain infectivity, and epistatic changes in one can permit inactivating mutations in the other. While both HA and NA functions are essential for infectivity and persistence and must be in balance, the mechanisms of functional cooperation or antagonism between them during either cellular entry or egress remain unexplained. Our data implicate both HA and NA functions in affecting virion-shape outcomes. We are using HA/NA coevolution as a paradigm to explore the role of virion shape heterogeneity in permitting viral adaptation to immune pressure.
Novel antivirals
In collaboration with the Fraden, Rogers, and Hagan groups at Brandeis, we are exploring an unconventional antiviral concept: rather than targeting viral proteins individually by small molecules or antibodies, our approach is to engulf whole virions within de novo designed macromolecular shells. The shells are made from DNA origami building blocks and use the same symmetry-based design principles that are used by icosahedral viral capsids. Antibodies attached on the inside of the shells (hundreds per shell) ensure specificity in assembly. The key advantage of this approach is that virus neutralization requires just one initiating capsomere to bind to the virus, yet the multivalent binding for the assembled shells leaves little room for escape by mutation that reduces antibody affinity. Engineering tubular shells will enable specific targeting of filamentous particles in a pleomorphic population. By engulfing whole virions, the built-in resistance of filamentous particles to inactivation of their individual glycoproteins will be eliminated. Complementary efforts are underway in the Ivanovic lab to develop antivirals that will interfere with filamentous virion assembly. We are also working to identify host factors required for cell entry by the filamentous virus particles to enable their specific targeting by broadly acting antivirals.

